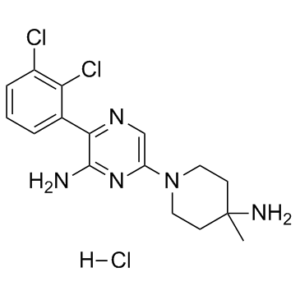
| In Vitro | In vitro activity: X-ray co-crystal of SHP-099 in complex with SHP2 revealed a new interaction with the basic amine and the Phe113 backbone carbonyl of the receptor. SHP099 shows inhibition of cell proliferation in the KYSE-520 model with an IC50 of 1.4 μM. SHP099 displayed high solubility and high permeability with no apparent efflux in Caco-2 cells. SHP099 concurrently binds to the interface of the N-terminal SH2, C-terminal SH2, and protein tyrosine phosphatase domains, thus inhibiting SHP2 activity through an allosteric mechanism. SHP099 suppresses RAS–ERK signalling to inhibit the proliferation of receptor-tyrosine-kinase-driven human cancer cells.
Kinase Assay: Biochemical assay. SHP2 is allosterically activated through binding of bis-tyrosylphorphorylated peptides to its Src Homology 2 (SH2) domains. The latter activation step leads to the release of the auto-inhibitory interface of SHP2, which in turn renders the SHP2 PTP active and available for substrate recognition and reaction catalysis. The catalytic activity of SHP2 was monitored using the surrogate substrate DiFMUP in a prompt fluorescence assay format. More specifically, the phosphatase reactions were performed at room temperature in 384-well black polystyrene plate, flat bottom, low flange, non-binding surface (Corning, Cat# 3575) using a final reaction volume of 25 μL and the following assay buffer conditions : 60 mM HEPES, pH 7.2, 75 mM NaCl, 75 mM KCl, 1 mM EDTA, 0.05% P-20, 5 mM DTT. The inhibition of SHP2 from the tested compounds (concentrations varying from 0.003 – 100 μM) was monitored using an assay in which 0.5 nM of SHP2 was incubated with of 0.5 μM of peptide IRS1_pY1172(dPEG8)pY1222(sequence:H2NLN(pY)IDLDLV(dPEG8)LST(pY)ASINFQK-amide). After 30-60 minutes incubation at 25 oC, the surrogate substrate DiFMUP (Invitrogen, cat# D6567, 200 μM) was added to the reaction and incubated at 25 oC for 30 minutes (200 μM for 2-593, 100 μM for 1-525 construct). The reaction was then quenched by the addition of 5 μL of a 160 μM solution of bpV(Phen) (Enzo Life Sciences cat# ALX-270-204). The fluorescence signal was monitored using a microplate reader (Envision, Perki-Elmer) using excitation and emission wavelengths of 340 nm and 450 nm, respectively. The inhibitor dose response curves were analyzed using normalized IC50 regression curve fitting with control based normalization.
Cell Assay: p-ERK cellular assay using the AlphaScreen(R) SureFire(TM) Phospho-ERK 1/2 Kit (PerkinElmer): KYSE-520 cells (30,000 cells/well) were grown in 96-well plate culture overnight and treated with SHP2 inhibitors at concentrations of 20, 6.6, 2.2, 0.74, 0.24, 0.08, 0.027 μM for 2 h at 37 °C. Incubations were terminated by addition of 30 μL of lysis buffer (PerkinElmer) supplied with the SureFire phospho-extracellular signal-regulated kinase (p-ERK) assay kit (PerkinElmer). Samples were processed according to the manufacturer's directions. The fluorescence signal from p-ERK was measured in duplicate using a 2101 multilabel reader (Perkin Elmer Envision). The percentage of inhibition was normalized by the total ERK signal and compared with the DMSO vehicle control. |
|---|
| In Vivo | SHP099 exhibits dose-dependent inhibition of tumor growth in xenograft models. After a single doses of 30 and 100 mg/kg, dose-dependent exposure and modulation of the pharmacodynamic marker p-ERK is observed in the xenografts. A daily oral dose of 10 or 30 mg/kg yield 19% and 61% tumor growth inhibition, respectively. Tumor stasis is achieved at 100 mg/kg. All animal studies were carried out according to the Novartis Guide for the Care and Use of Laboratory Animals. Female nude mice were inoculated subcutaneously (3 x 106 cells) in a suspension containing 50% phenol red-free matrigel (BD Biosciences) in Hank’s balanced salt solution with parental KYSE-520 cells. For PK/PD studies, mice were administered a single dose of vehicle control or 1 by oral gavage once tumors reached roughly 500 mm3. Mice were subsequently euthanized at predetermined time points following a single dose of compound at which point plasma and xenograft fragments were harvested for determination of 1 concentrations and p-ERK modulation, respectively. For efficacy studies, mice were calipered twice weekly by calipering in two dimensions. Once tumors reached roughly 200 mm3, mice were randomly assigned to treatment groups. For the efficacy study, mice were assigned to receive either vehicle, 1 (10, 30, or 100 mg/kg qd), or erlotinib (80 mg/kg qd) by oral gavage. Tumor volume and mouse body weight was assessed twice weekly. To assess MAPK pathway modulation in xenograft protein lysates, total and phospho-ERK1/2 was assessed using a commercially available kit (Meso Scale Discovery catalog number K15107D). The assay was conducted as recommended by Meso Scale Discovery with the exception that protein lysate was incubated overnight. |
|---|