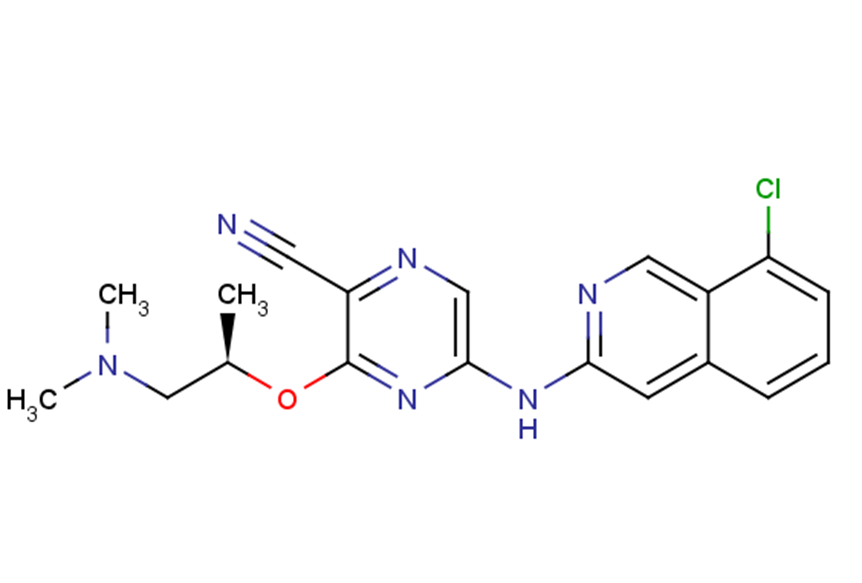
SAR-020106 是一种强效、ATP 竞争性和选择性 Chk1 抑制剂,IC50为 13.3 nM。它对 Chk2 具有良好的选择性,可通过选择抗癌药物增强抗肿瘤活性。
产品描述
SAR-020106 is a potent, ATP-competitive, and selective CHK1 inhibitor with an IC50 of 13.3 nmol/L on the isolated human enzyme.
体外活性
SAR-020106 potentiated the efficacies of irinotecan and gemcitabine in SW620 human colon carcinoma xenografts in nude mice[2]
体内活性
SAR-020106 is an ATP-competitive, potent, and selective CHK1 inhibitor with an IC(50) of 13.3 nmol/L on the isolated human enzyme. This compound abrogates an etoposide-induced G(2) arrest with an IC(50) of 55 nmol/L in HT29 cells, and significantly enhances the cell killing of gemcitabine and SN38 by 3.0- to 29-fold in several colon tumor lines in vitro and in a p53-dependent fashion. Biomarker studies have shown that SAR-020106 inhibits cytotoxic drug-induced autophosphorylation of CHK1 at S296 and blocks the phosphorylation of CDK1 at Y15 in a dose-dependent fashion both in vitro and in vivo. Cytotoxic drug combinations were associated with increased gammaH2AX and poly ADP ribose polymerase cleavage consistent with the SAR-020106-enhanced DNA damage and tumor cell death. Irinotecan and gemcitabine antitumor activity was enhanced by SAR-020106 in vivo with minimal toxicity. SAR-020106 represents a novel class of CHK1 inhibitors that can enhance antitumor activity with selected anticancer drugs in vivo and may therefore have clinical utility[1].
Cas No.
1184843-57-9
分子式
C19H19ClN6O
分子量
382.85
储存和溶解度
DMSO:10 mM
Powder: -20°C for 3 years
In solvent: -80°C for 2 years