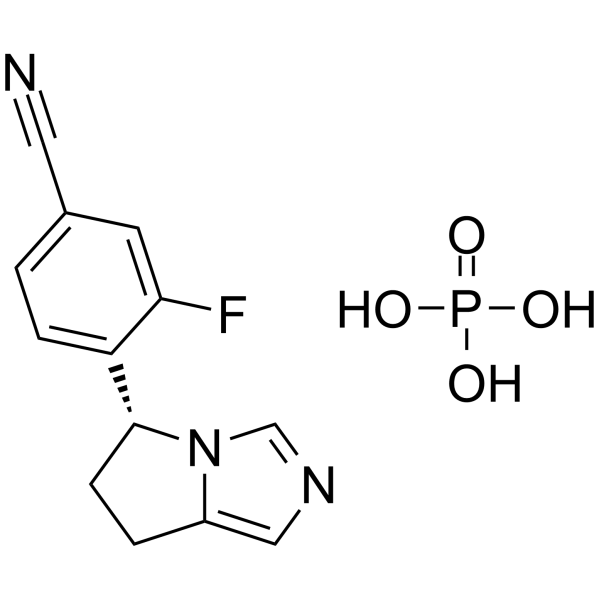
Osilodrostat (LCI699) phosphate 是一种口服有效的 11β-羟化酶 (CYP11B1) 抑制剂,其 IC50 值为 35 nM。Osilodrostat phosphate 是一种口服有效的醛固酮合成酶 (CYP11B2) 抑制剂,抑制人醛固酮合成酶和大鼠醛固酮合成酶的 IC50 分别为 0.7 nM 和 160 nM。Osilodrostat phosphate 抑制醛固酮和皮质酮的合成。Osilodrostat phosphate 具有降低血压的能力。Osilodrostat phosphate 可用于库欣综合征 (CS) 的研究。
| Cas No. | 1315449-72-9 |
| 别名 | LCI699 phosphate |
| 分子式 | C13H13FN3O4P |
| 分子量 | 325.23 |
| 溶解度 | H2O : ≥ 200 mg/mL (614.95 mM) |
| 储存条件 | 4°C, away from moisture |
| General tips | For obtaining a higher solubility , please warm the tube at 37 ℃ and shake it in the ultrasonic bath for a while. |
| Shipping Condition | Evaluation sample solution : ship with blue ice
All other available size: ship with RT , or blue ice upon request |
| 产品描述 | IC50: 35 nM (CYP11B1), 0.7 nM (human aldosterone synthase), and 160 nM (rat aldosterone synthase)[1][2] Osilodrostat (LCI699) phosphate is a potent, orallyactive11β-hydroxylase (CYP11B1)inhibitor with anIC50value of 35 nM. Osilodrostat phosphate is a potent, orallyaldosterone synthase (CYP11B2)inhibitor withIC50values of 0.7 nM and 160 nM for human aldosterone synthase and rat aldosterone synthase, respectively. Osilodrostat phosphate inhibits aldosterone and corticosterone synthesis. Osilodrostat phosphate has blood pressure lowering ability. Osilodrostat phosphate can be used for research of Cushing syndrome (CS)[1][2][3]. Osilodrostat (LCI699; 0.01-10 µM; HAC15 cells, 17 primary human adrenocortical cell cultures, and pituitary adenoma cells) phosphate inhibits cortisol and aldosterone. Osilodrostat results in inhibition of corticosterone, 11-deoxycortisol accumulation, and modest effects on adrenal androgens[2]. Osilodrostat (LCI699; 0.1-100 mg/kg; p.o.; once) phosphate inhibits aldosterone and corticosterone synthesis in Ang-II- and ACTH-stimulated Sprague Dawley rats[1].
Osilodrostat (LCI699; 3-100 mg/kg; p.o.; daily, for 52 weeks) phosphate reduces mean arterial pressure and prolongs survival in dTG rats[1]. | Animal Model: | Male Ang-II- and ACTH-stimulated Sprague Dawley rats[1] | | Dosage: | 0.1, 0.3, 1 and 3 mg/kg (Ang-II-stimulated rats) and 1, 3, 10, 30 and 100 mg/kg (ACTH-stimulated rats) | | Administration: | Oral administration; once | | Result: | Inhibited the increase in plasma aldosterone concentrations stimulated by Ang II or ACTH in a dose-dependent manner. |
| Animal Model: | dTG rats[1] | | Dosage: | 3, 10, 30 and 100 mg/kg | | Administration: | Oral administration; daily, for 52 weeks | | Result: | Increased fractional LV (systolic and diastolic) shortening, normalized LV isovolumic relaxation time to RR (IVRT/RR) ratio and myocardial cell size and reduced LV weight in a dose-dependent manner. |
|