
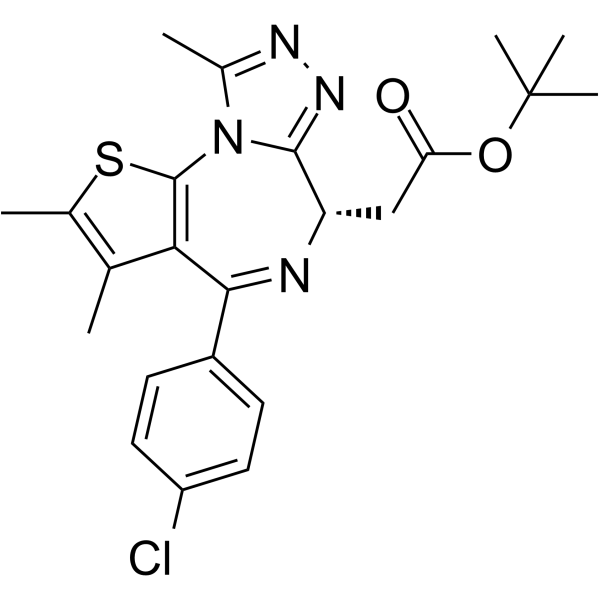
| CAS NO: | 1268524-70-4 |
| 生物活性 | (+)-JQ-1 (JQ1) is a potent, specific, and reversibleBET bromodomaininhibitor, withIC50s of 77 and 33 nM for the first and second bromodomain (BRD4(1/2))[1]. (+)-JQ-1 also activatesautophagy[2]. | ||||||||||||||||
| IC50& Target | IC50: 77/33 nM (BRD4(1/2))[1] | ||||||||||||||||
| 体外研究 (In Vitro) | (+)-JQ-1 represents a potent, highly specific and Kac competitive inhibitor for the BET family of bromodomains. (+)-JQ-1 (100 nM, 48 h) prompts squamous differentiation exhibited by cell spindling, flattening and increased expression of keratin. (+)-JQ-1 (250 nM) induces rapid expression of keratin in treated NMC 797 cells compared to (-)-JQ1 (250 nM) and vehicle controls, as determined by quantitative immunohistochemistry.(+)-JQ-1 (250 nM) elicits a time-dependent induction of strong (3+) keratin staining of treated NMC 797 cells, compared to (-)-JQ1 (250 nM)[1]. De-repression of autophagy genes is observed almost immediately after (+)-JQ-1 addition[2]. (+)-JQ-1 is a potent thienodiazepine inhibitor (Kd=90 nM) of the BET family coactivator protein BRD4, which is implicated in the pathogenesis of cancer via transcriptional control of the MYC oncogene. Dose-ranging studies of (+)-JQ-1 demonstrates potent inhibition of H4Kac4 binding with a IC50value of 10 nM for murine BRDT(1) and 11 nM for human BRDT(1)[3]. | ||||||||||||||||
| 体内研究 (In Vivo) | Matched cohorts of mice with established tumors are randomized to treatment with (+)-JQ1 (50 mg/kg) or vehicle, administered by daily intraperitoneal injection. Prior to randomization, and after four days of therapy, mice are evaluated by FDG-PET imaging. A marked reduction in FDG uptake is observed with (+)-JQ1 treatment. Tumor-volume measurements confirm a reduction in tumor growth with JQ1 treatment. Pharmacokinetic studies of (+)-JQ1 are performed in CD1 mice following intravenous and oral administration. Mean plasma concentration-time profiles of (+)-JQ1 after intravenous dosing (5 mg/kg). The pharmacokinetic parameters for intravenous (+)-JQ1 demonstrate excellent drug exposure (AUC=2090 hr*ng/mL) and an approximately one hour half-life (T1/2). Mean plasma concentration-time profiles of (+)-JQ1 after oral dosing (10 mg/kg). The pharmacokinetic parameters for oral (+)-JQ1 demonstrate excellent oral bioavailability (F=49%), peak plasma concentration (Cmax=1180 ng/mL) and drug exposure (AUC=2090 hr*ng/mL)[1]. | ||||||||||||||||
| 分子量 | 456.99 | ||||||||||||||||
| 性状 | Solid | ||||||||||||||||
| Formula | C23H25ClN4O2S | ||||||||||||||||
| CAS 号 | 1268524-70-4 | ||||||||||||||||
| 运输条件 | Room temperature in continental US; may vary elsewhere. | ||||||||||||||||
| 储存方式 |
| ||||||||||||||||
| 溶解性数据 | In Vitro: DMSO : ≥ 45 mg/mL(98.47 mM) *"≥" means soluble, but saturation unknown. 配制储备液
* 请根据产品在不同溶剂中的溶解度选择合适的溶剂配制储备液;一旦配成溶液,请分装保存,避免反复冻融造成的产品失效。 In Vivo: 请根据您的实验动物和给药方式选择适当的溶解方案。以下溶解方案都请先按照In Vitro方式配制澄清的储备液,再依次添加助溶剂: ——为保证实验结果的可靠性,澄清的储备液可以根据储存条件,适当保存;体内实验的工作液,建议您现用现配,当天使用;
以下溶剂前显示的百
|
| 维奥蛋白资源库 - 中文蛋白资源 CopyRight © 2010-2026 |